03. ABRYSVO:
ADULTO MAYOR O IGUAL A 60 AÑOS
El 30 de septiembre de 2022, Pfizer, presentó una solicitud de Licencia de Productos Biológicos a la FDA de la vacuna RSVpreF: Abrysvo® para prevenir la enfermedad respiratoria aguda y la enfermedad del tracto respiratorio inferior causada por VSR en personas de 60 años o más. Se trata de una vacuna bivalente de subunidad proteica estabilizada recombinante y consiste en cantidades iguales de antígenos F en prefusión de los dos subgrupos principales del VRS: prefusión F del subgrupo A del VRS (60 µg) y prefusión F del subgrupo B del VRS (60 µg). El esquema propuesto es una inyección intramuscular única de 120 µg por dosis.
Se presentaron datos de 6 estudios clínicos, los datos que respaldan la seguridad y eficacia de RSVpreF en personas de 60 años o más provienen de un ensayo multinacional de fase 3, aleatorizado, doble ciego y controlado con placebo (Estudio C3671013 o Estudio 1013). Un total de 34.284 participantes que fueron asignados al azar para recibir una dosis única de RSVpreF (n=17.215) o placebo (n=17.069). Se realizó un análisis provisional especificado en el protocolo que evaluó eficacia para prevenir la enfermedad del tracto respiratorio inferior asociada al VRS (LRTI-RSV) confirmada por laboratorio con ≥2 síntomas y ≥3 síntomas con inicio al menos 14 días después de la vacunación. A partir del corte de datos del 8 de julio de 2022, la duración media del seguimiento de la eficacia fue de aproximadamente 7 meses.
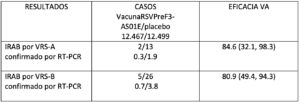
La eficacia de la vacuna (EV) para prevenir las LRTI-RSV confirmadas por laboratorio con ≥2 síntomas fue del 66,7 % (IC 96,66%: 28,8; 85,8), 11 casos en el grupo vacunado y 33 casos en el grupo placebo. La EV en la prevención de LRTI-RSV confirmada por laboratorio con ≥3 síntomas fue del 85,7 % (IC 96,66%: 32,0; 98,7), 2 casos en el grupo vacunado y 14 casos en el grupo placebo. La EV contra la infección respiratoria aguda asociada al VRS (IRA-RSV) demostró ser del 62,1 % (IC 95%: 37,1, 77,9).
Si bien la eficacia de la vacuna parece conservarse entre los participantes ≥80 años y entre los participantes con al menos un factor de riesgo de VSR grave, la interpretación está limitada por el pequeño tamaño de la muestra y el bajo número de casos para estos subgrupos.

Actualmente no hay datos disponibles sobre la duración de la efectividad; EV en inmunocomprometidos y en adultos mayores frágiles, eficacia en la prevención de casos graves de IRAB. Existen datos limitados sobre la EV en individuos ≥80 años de edad. Tampoco hay datos disponibles sobre la duración de la respuesta inmune y la seguridad e inmunogenicidad de la administración concomitante con vacunas recomendadas para uso rutinario en esta población.
En cuanto a seguridad, el estudio 1013 incluyó a 34.284 participantes vacunados, 26.395 (77,0%) tuvieron al menos 6 meses de seguimiento después de la vacunación. Los datos sobre reacciones adversas (RA) locales y sistémicas solicitadas dentro de los 7 días posteriores a la vacunación se recopilaron de un subconjunto de 7.196 participantes del estudio. Las RA de mayor frecuencia (>10%) fueron fatiga (15,5%), dolor de cabeza (12,8%), dolor en sitio de inyección (10,6%) y dolor muscular (10,1%); en su mayoría leves y moderadas, con un 0,2 % y un 0,7 % notificadas como de grado 3 respectivamente. En general, las RA solicitadas se informaron con mayor frecuencia en el subgrupo de edad más joven (60-69 años) en comparación con los subgrupos de mayor edad. La mayoría de los AR solicitados se resolvieron entre 1 y 2 días después de la vacunación.
La FDA consideró que tres RA graves, estaban posiblemente relacionados con la vacuna: un evento de hipersensibilidad, no clasificado como anafilaxia, que comenzó 8 horas después de la vacunación; un caso de síndrome de Guillain-Barré (SGB) que comenzó 7 días después de la vacunación; y un caso de síndrome de Miller Fisher que comenzó 8 días después de la vacunación. Dada la relación incierta entre RSVpreF y SGB (incluido MFS) y otras afecciones desmielinizantes inmunomediadas, la FDA recomienda al fabricante presentar una propuesta para un estudio de seguridad post comercialización para evaluar el riesgo de GBS y otras afecciones desmielinizantes inmunomediadas entre las personas vacunadas con RSVpreF.[1]
Se produjeron 52 muertes (0,3%) de los receptores de RSVpreF y en 49 (0,3%) de los receptores de placebo. Ninguna de las muertes se consideró relacionada con la intervención del estudio.
El 31 de mayo del 2023 el Comité Asesor de Vacunas y Productos Biológicos Relacionados de los Estados Unidos (VRBPAC), ha recomendado la autorización de la comercialización de la vacuna Abrysvo® de Pfizer para la prevención de las infecciones respiratorias bajas causadas por el VRS con la indicación de uso en los adultos de sesenta o más años.
EMBARAZADAS:
El estudio de fase 3 NCT04424316 se incluyó sitios en países del hemisferio norte y sur, se evaluó la eficacia para prevenir la enfermedad del tracto respiratorio inferior (LRTD) asociada al VRS en recién nacidos de embarazadas vacunadas. El estudio evaluó la eficacia para prevenir la LRTD asociada al VRS y LRTD grave asociada al VRS en los lactantes dentro de los 90, 120, 150 y 180 días después del nacimiento. Las participantes fueron asignadas al azar (1:1) para recibir 1 dosis de vacuna o placebo. La eficacia de la vacuna (EV) se definió como la reducción del riesgo relativo de los criterios de valoración de LRTD grave causada por VRS y LRTD causada por VRS en recién nacidos de embarazadas vacunadas en comparación con recién nacidos de embarazadas que recibieron placebo.
Las participantes fueron asignadas al azar (1:1) para recibir vacuna (3.695) o placebo (3.697). La LRTD asociada al VRS se definió como una LRTD con reacción en cadena de polimerasa con transcripción inversa (RT-PCR) que requirió visita médica con uno o más de los siguientes síntomas respiratorios: taquipnea (frecuencia respiratoria ≥60 respiraciones/minuto en < 2 meses de edad, ≥50 en ≥2 a 12 meses de edad], o ≥40 en ≥12-24 meses de edad); Saturación de O2 (SpO2) medida en aire ambiente <95%; tiraje de la pared torácica. La LRTD grave asociada al VSR fue definida como aquel que cumplía los criterios del LRTD RSV más al menos uno de los siguientes: taquipnea (frecuencia respiratoria ≥70 respiraciones por minuto en <2 meses de edad, ≥60 en ≥2 a 12 meses de edad, o ≥50 en ≥12 a 24 meses de edad); SpO2 medida en aire ambiente <93 %; cánula nasal de alto flujo o ventilación mecánica (invasiva o no invasiva), ingreso en UCI >4 horas y/o falta de respuesta/inconsciencia. Los criterios de valoración secundarios de eficacia incluyeron las hospitalizaciones debidas al VRS.
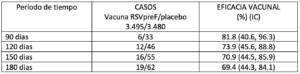
Los resultados de EV cumplieron el criterio estadístico de éxito (límite inferior IC >20 %) para reducir la LRTD grave debida al VRS, dentro de los 180 días. Los resultados de EV no cumplieron con el criterio estadístico de éxito (límite inferior IC >20%) para reducir la LRTD debida al VRS; sin embargo, se observó una eficacia clínicamente significativa entre 90 y 180 días después del nacimiento.
En el estudio NCT04424316 se excluyeron embarazadas con embarazos de alto riesgo (IMC>40 kg/m2 antes del embarazo, embarazos resultantes de fertilización in vitro, preeclampsia, eclampsia, hipertensión gestacional no controlada, anomalías placentarias, polihidramnios u oligohidramnios, sangrado significativo o trastorno de coagulación) trastorno endocrino inestable (intolerancia a la glucosa no tratados o trastornos de la tiroides). Se podían incluir embarazadas con complicaciones previas del embarazo (antecedentes de parto prematuro ≤ 34 semanas de gestación, muerte fetal previa, muerte neonatal, recién nacido previo con un trastorno genético conocido o anomalía congénita significativa.
La mediana de edad materna en el momento de la vacunación fue de 29 años (16 a 45 años en el grupo vacuna, de 14 a 47 años en el grupo placebo). La mediana de edad gestacional en el momento de la vacunación fue de 31 semanas y 2 días (rango 24-36,9 semanas).
La mayoría de las reacciones locales y sistémicas solicitadas en las madres participantes se resolvieron dentro de los 2 o 3 días posteriores. Se informaron reacciones locales graves en el 0,3 % de las participantes en el grupo vacuna y ninguna en el grupo placebo, y el 2,3% de las participantes en ambos grupos informaron reacciones sistémicas graves dentro de los 7 días posteriores a la vacunación.
Los eventos adversos no solicitados informados dentro del mes posterior a la vacunación fueron del 13,7 % en el grupo de vacuna y del 13,1 % en el grupo de placebo. Los eventos informados con más frecuencia en las madres participantes fueron trastornos del embarazo, puerperio y condiciones perinatales (7,0% para el grupo vacuna vs 6,2% para el grupo placebo).
Los eventos adversos graves que ocurrieron en cualquier momento durante el estudio fueron informados por un 16,2% en el grupo de vacuna y un 15,2% en el grupo de placebo, con un 4,2% de eventos adversos graves dentro de 1 mes después de la vacunación en el grupo vacunado y un 3,7% en el grupo placebo. La mayoría de los eventos adversos graves en las madres participantes se relacionaron con complicaciones del embarazo y ocurrieron después del mes posterior a la vacunación.
Se observó un desequilibrio numérico en los nacimientos prematuros en las receptoras de vacuna en comparación con las que recibieron placebo en los Estudios 1 y 2. En el Estudio 2, los nacimientos prematuros ocurrieron en el 5,3% (6 de 114) en el grupo vacunado y en el 2,6% (3 de 116) en el grupo placebo. En el Estudio 1 los partos prematuros ocurrieron en el 5,7% ([IC 95%: 4,9, 6,5] (202 de 3.568)) en el grupo vacunado y en el 4,7% ([IC 95%: 4,1, 5,5] (169 de 3.558)) en el grupo placebo.
En el Estudio 1, se observaron eventos adversos desde el nacimiento hasta 1 mes de edad en el 37,1% del grupo vacuna en comparación con el 34,5% del grupo placebo. Se observó bajo peso al nacer en el 5,1 % del grupo de madre3s vacunadas frente al 4,4% del grupo de placebo, y se observó ictericia neonatal en el 7,2% del grupo vacuna frente al 6,7% del grupo de placebo.
Los nacidos en el año 1 tendrán un seguimiento de hasta 24 meses y los nacidos en el año 2 serán seguidos durante un máximo de 12 meses para evaluar la seguridad. En el momento de la evaluación de los datos, después de una mediana de 8,9 meses, habían nacido 3.568 hijos de madres del grupo vacunado y 3.558 del grupo placebo, y de estos, aproximadamente el 45,6 % habían sido seguidos durante 12 meses.
El 21 de agosto del 2023 el Comité Asesor de Vacunas y Productos Biológicos Relacionados de los Estados Unidos (VRBPAC), ha recomendado la autorización de la comercialización de la vacuna Abrysvo® de Pfizer para la inmunización activa de embarazadas de 32-36S de EG para la prevención LRTD y LRTD grave por VRS en niños desde RN hasta los 6 meses de edad.